Quick links
Third party software
Links
iSeq News
- 08/03/2017: We have released hygestat_bless-1.3.3: several bugs reported by users have been corrected
- 06/13/2017: We have released hygestat_bless-1.3.2: several bugs reported by users have been corrected
- 02/20/2017: A full documentation on how to use iSeq for the analysis and interpretation of DSB-Sequencing data is now available online.
- 12/09/2016: We have released hygestat_bless-1.3: several bugs reported by users have been corrected
- 03/27/2016: hygestat_bless now support all sequencing technologies and systems
- 02/15/2016: hygestat_bless now support Bowtie2 and zipped files as input
Tutorial on how to use iSeq software to reproduce our results from Crosetto et al, Nature Methods 10, 361–365 (2013) is coming soon.
How to download and install iSeq suite of software.
This section describes how to download the iSeq suite of software. We also explain how to install hygestat_BLESS, the main component of iSeq. The Tutorial section contains information on how to install and use additional modules: hygestat_genes, hygestat_annotation, find_gene and the Matlab visualization procedure. Hygestat_BLESS is the core part of the iSeq software that identifies regions statistically significantly enriched in DNA double stranded breaks using DSB sequencing data such as BLESS. The software is compatible with modern versions of Fedora and Kubuntu (e.g. Fedora 32). Please download the following files:| File | Type | Description |
|---|---|---|
| hygestat_bless-1.2.4.tar.gz | Required | Stable version of hygestat_bless, core component of iSeq |
| hygestat_annotation.1.0.tar.gz | Optional | hygestat_annotation compares enriched genomic regions found by hygestat_BLESS with with user-selected genome annotation data |
| Hygestat_bed.1.0.tar.gz | Optional | hygestat_bed computes probabilities that the genomic regions provided by the user are significantly enriched in DSBs. |
| Hygestat_genes.1.0.tar.gz | Optional | hygestat_genes computes probabilities for all in the considered organism to be statistically enriched in the treatment sample reads (DSBs for the BLESS data), same probabilities are computed for gene promoters and UTRs. |
| Hygestat_plots.1.0.tar.gz | Optional | hygestat_plots is a set of Matlab scripts to visualize iSeq results. |
| Hygestat_mappability.1.0.tar.gz | Optional | hygestat_mappability is a set of scripts to compute human, mouse and yeast mappability. |
| mouse_mappability_45.tar.gz | Optional | Mouse genome mappability |
| human_mappability_45.tar.gz | Optional | Human genome mappability |
| sample_configuration_file.txt | Optional | Sample configuration file for hygestat_bless |
| yeast_mappability_45.tar.gz | Optional | Yeast genome mappability |
| countReads | Optional | Count the number of reads in a specific region (eg. G-quadruplex) |
| supplementary_files.tar.gz | Optional | All supplementary files |
How to install hygestat_bless, hygestat_genes, hygestat_bed and find_gene
- Download hygestat_bless from the link above or here
- Decompress the file $ tar xvzf hygestat_bless-xxx.tar.gz
- Go to the main directory $ cd hygestat_bless-xxx
- Run the configuration script $ ./configure [option]
- If bowtie is not in your PATH (check with the command $ which bowtie): run $ ./configure --with-bowtie=/path/to/bowtie/ (step 4)
- To install hygestat_BLESS with fastqc: run $ ./configure --with-fastqc=/path/to/fastqc/ (step 4)
- To install hygestat_BLESS with samstat: run $ ./configure --with-samstat=/path/to/samstat/ (step 4)
- Example: installation with bowtie, fastqc and samstat : run $ ./configure --with-bowtie=/path/to/bowtie/ --with-fastqc=/path/to/fastqc/ --with-samstat=/path/to/samstat/
- If you do not have administration privileges on your computer: run $ ./configure --prefix=/path/to/install/ , then $ export PATH=$PATH:/path/to/install/
- Run the Makefile $ make
- Proceed to the installation (as root) $ make install
- If successfuly installed open the help menu of hygestat_BLESS $ hygestat_bless --help
How to run hygestat_BLESS
hygestat_BLESS currently supports two types of usage:- hygestat_bless -i config_file.txt [default] config_file.txt : configuration file that contains all the parameters (example can be downloaded here). This is the recommended option as many parameters can be kept as default.
- hygestat_bless [options] control.[fastq/zip] treatment.[fastq/zip]
In this option, all parameters are specified in the command line (expand table for the list of options).
Option Extended Option Parameter Description Default Behavior -o --output-dir <hygest-results> write all output files to this directory [ default: ./ ] -O --output-file <output.txt> main output file name [ default: output.txt ] -p --num-threads <1> number of threads used during analysis [ default: 1 ] -G --genomeDir </path/to/genome> absolute directory path to the reference genome [ default: ./ ] -g --genomeType <genome> genome type (currently support mouse human and yeast genome) [no default: ] -F --fastqControl <control.fastq> control fastq file [ no default: ] -N --natureControl <bless/genomic> nature of the control data [ default: genomic] -f --fastqTreatment <treatment.fastq> treatment fastq file [ no default: ] -n --natureTreatment <bless/genomic> nature of the treatment data [ default: bless] -d --dataDir </path/to/fastq/files> absolute directory path to the data [ default: ./ ] -m --mapDir </path/to/mappability/files> absolute directory path to the data [ default:no mappability] -b --noBowtie <FALSE/TRUE> skip alignement with bowtie [ default: FALSE ] -t --telomere <FALSE/TRUE> estimate telomeres contribution to the analysis [ default: FALSE ] -s --time-serie <FALSE/TRUE> time serie analysis [provide a configuration file] [ default: FALSE ] --time-serie-file <config_time_serie.txt> time serie configuration file [no default: ] --time-serie-rare <FALSE/TRUE> rarefaction correction of time serie data [ default: FALSE ] --time-serie-rand <20> number of rarefaction data [will use number +1] [ default: 20] -a --preCheck <FALSE/TRUE> use FastQC for pre-quality check of all fastq files [ default: FALSE] -k --wigOutput output wig files for visualization [ default: yes ] -A --postCheck <FALSE/TRUE> use samstat for post alignment analysis [ default: FALSE] -r --resolution <10250> resolution of the dsb detection [ default: 10250] -w --windowsAdvance <1> windows advance [ default: 1] -c --correction <false/true> run bless with correction [ default: false ] --corTreatment <correction_treatment.fastq> treatment sample for correction [ no default: ] --corTreatmentNat <bless/genomic> nature of the correction treatment [ no default: ] --corControl <correction_control.fastq> control sample for correction [ no default: ] --corControlNat <bless/genomic> nature of the correction control [ no default: ] --corFibroblast <correction_fibroblast.fastq> fibroblast [ no default: ] --corFibroblastNat <bless/genomic> nature of fibroblast [ no default: ] -y --cytoPath <cytoband-human.txt> absolute path to the cytoband file [ no default: ] -Y --fragilePath <fragile-band-human.txt> absolute path to the fragile band file [ no default: ] -v --verbose log-friendly verbose processing (no progress bar) [ default: FALSE ] -q --quiet log-friendly quiet processing (no progress bar) [ default: FALSE ] -h --help print help menu [ no default: ] --no-update-check do not contact server to check for update availability [ default: FALSE ]
Documentation: Identifying and annotating regions significantly enriched in breaks in DSB sequencing data.
1 - MATERIALS
1 - 1 - Equipment
- Computer running with Linux operating system with a minimum setup, see equipment setup.
- DNA sequencing data, such as BLESS, or ChIP-Seq
- Newest version of iSeq, at least core procedure, hygestat_BLESS, that can be downloaded here (download hygestat_BLESS) and all third party software as described in equipment setup section.
1 - 2 - Equipment setup
- Hardware requirement and computer configuration: iSeq is a software pipeline, written mainly in C++, with some additional python, perl and shell scripts. The latest version of C++(gcc) compiler should be installed (generally comes with linux operating system, just needs to be updated), C++ libraries called boost ( download boost) and gsl ( download gsl ) have to be installed as well. hygestat_bless uses bowtie or bowtie2 to align the reads to the reference genome (bowtie is used as default). For information about how to install bowtie and bowtie build index, go to bowtie website (download bowtie ). hygestat_bless optionally uses samstat and/or fastqc (download samstat, download fastqc) to preprocess and check the quality of the data before analysis. A computer (64-bit), running in linux with a minimum of 2 GB of RAM, 100 GB of free space in hard drive (HD) is required to complete this tutorial.
- Input data:
hygestat_bless currently accepts data in fastq or fasta format from any NGS pipeline (Illumina, Roche 454 etc).
Data can be genomic (here we consider genomic all data without a barcode) or BLESS (data with specific sequencing barcode, that can be user-defined).
In this tutorial, we will use actual BLESS barcodes, but the procedure can be easily adapted to any type of barcoded data.
Data used in this tutorial can be obtained from NCBI Sequence Read Archive
(accession number SRP018506) or
from our website breakome.
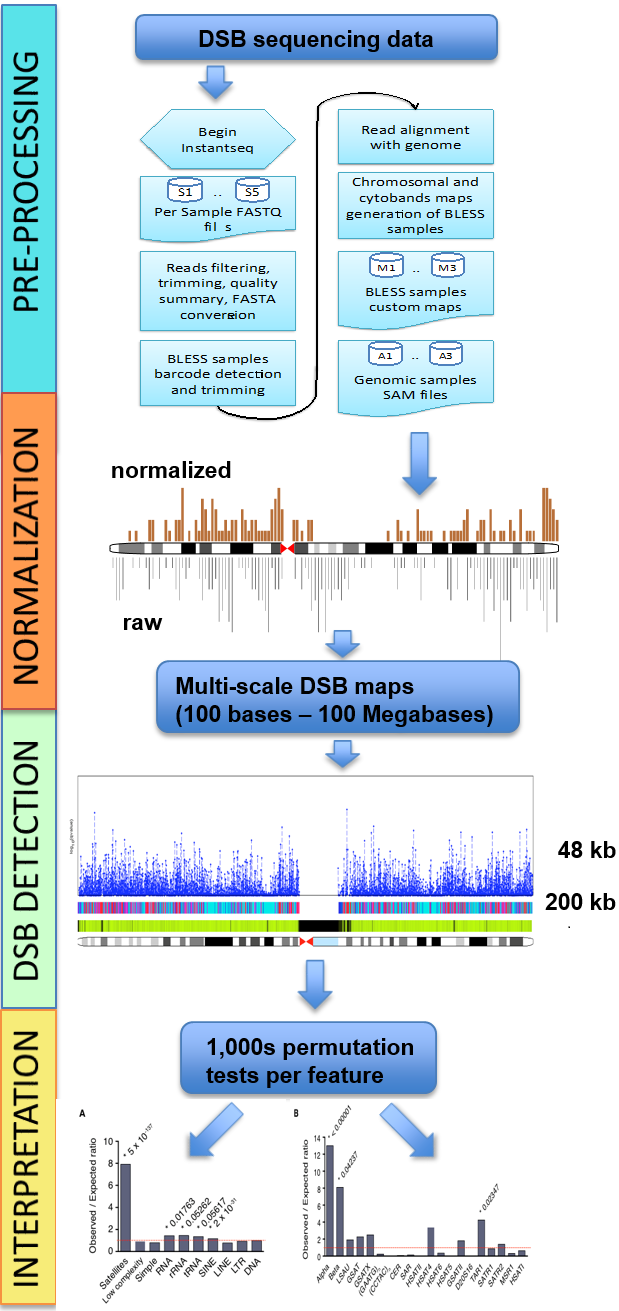
Once all data and requirements have been successfully setted up, no action is needed until the end of the process with the output file written in output.txt. - Output data: containing 8 columns. These columns are, starting from the first: chromosome number, window start position, window end position, number of reads in sample of interest (treated sample) for the current window, number of reads in control (untreated sample) for the current window, p-value (calculated using hypergeometric test), q-value1 (p-value corrected for multiple hypothesis testing using Benjamini-Hochberg correction) and q-value2 (p-value corrected for multiple hypothesis testing using Bonferroni correction). An example of output file file is shown in the Table 1 bellow.
2 - PROCEDURE
2 - 1 - Overview
- Reading input data
- Reading input data and extracting sequence reads (in case of fastq data input).
- Pre-processing: Data trimming (optional) and barcode removal (if barcode used).
- Aligning to the reference genome using bowtie (download bowtie if not yet installed).
- Loading the pre-computed mappability map for the reference genome.
- Reading bowtie output (using provided hit.sh shell script) to produce the mapped positions for each chromosome and for each strand, saved in the directory called input_XX_barcode where XX stands for close or no barcode depending on point 3.
- Dividing genome into intervals of user-determined length in mappable reads (the intervals that, due to low mappability, would be more than 10 times longer than the user-provided target length are skipped and the procedure is started anew)
- Finding intervals (defined as in point 6) enriched in reads in the treated sample, using hypergeometric test (p-value is calculated separately for each window).
- alculating Benjamini-Hochberg or Bonferroni correction to correct p-values for multiple hypothesis testing (user can choose; Bonferroni correction is more rigorous than the Benjamini-Hochberg method and recommended for very deeply sequenced data sets, such as our recent yeast data (Biernacka et al. (2017), in preparation; Zhu et al., (2017A), in preparation); Zhu et al., (2017B), in preparation);.
- Creating an output table (eight column table (column explanation in the Table (add link) below) containing the results for each interval considered. For DSB sequencing data, the p-value indicates whether the given region is fragile (statistically significantly enriched in DNA double-stranded breaks).
- Annotating the results: hygestat_annotation automatically finds annotations enriched in significant regions found by hygestat_BLESS, using annotations from UCSC and NCBI, or user-provided.
- Identifying genes with significantly higher than expected numbers of reads in the treated sample (for BLESS, genes most prone to breaks) by computing the hypergeometric test for every gene (hygestat_genes software).
- Estimating probability for user-defined regions (in bed format) having significant enrichment of reads in the treated sample (hygestat_genes software).
- Analyzing locations of significantly fragile intervals with respect to transcription start and end sites (find_gene software).
- 14- Visualizing data on the chromosomes using the custom Matlab procedure. The pipeline software for visualization is available at the download section of this page, follow the steps in the README file to create the plots.
2 - 2- hygestat_BLESS critical steps
- Reading raw data and sequence read extraction. After successfully installing or compiling hygestat_BLESS (breakome), there are two possibilities to input the data: via command line or by using the config file, downloadable frombreakome. For more information about how to input the data and options of the software, please refer to our website (breakome)) and follow the instructions provided. In case of fastq data, the process_fastq function allows extraction of reads from fastq and produces the input.csv file containing information about the quality of the data. Troubleshooting: see troubleshooting section below.
- Pre-processing: data trimming and barcode removal. In the case of barcoded data, the shell script hit.sh allows removal of the barcode before any mapping to the reference genome. The script assumes that barcodes are located only at the beginning of reads and will search for reads that start with the user-provided barcode.
- Aligning to the reference genome. Reads are aligned to the reference genome using the third party software (bowtie). For more information about read alignment using bowtie, refer to the bowtie webpage (bowtie ).
- Loading the pre-computed mappability map for the reference genome. To take into account genome mappability during analysis, the iSeq software uses the pre-computed genome mappability files, that are available from our website for human, mouse and yeast, or can be computed for other organisms using our mappability software package, also available from our website (click here).
- Reading bowtie output The provided shell script hit.sh is used to read bowtie output and extract read positions mapped to the reference genome.
- Dividing genome into intervals of constant length in mappable nucleotides (user determined length). To reduce data complexity for further analysis, especially for annotating with known genomic features, we divide the genome into windows of equal length in mappable nucleotides, with window size defined by the user. If the actual size of the resulting window is 10-fold or bigger than user-defined (that is if the mappability is less than 10%), such window is skipped and the process is repeated from the beginning. We recommend analyzing data at the minimum of several different resolutions for spotting trends and overall more accurate results.
- Finding enriched intervals The hypergeometric statistical test is used here to evaluate the probability for a given interval of being significantly enriched in treatment sample reads (i.e. being “fragile”, in case of DSB sequencing data).
- Multiple hypothesis testing corrections Currently, two corrections for multiple hypothesis testing are supported: Benjamini-Hochberg correction (q-value #1) and Bonferroni correction (q-value #2). Bonferroni correction is more rigorous than Benjamini-Hochberg and is recommended for deeply sequenced samples, for example all the yeast samples. In our experience, for human and mouse samples Benjamini-Hochberg correction still gives better results, although it may change with increased depth of sequencing. Negative controls are very helpful in estimating the false positive ratio.
- Interpretation of the hygestat_BLESS output table
Table 1 below shows an example of a hygestat_BLESS output (named output.txt (default) or provided_name.txt). The output table consists of eight columns, with the following content:
- Column 1: Chromosome number (chrXX).
- Column 2: Chromosomal coordinate of window start.
- Column 3: Chromosomal coordinate of window end (note that depending on mappability, the size of the window in nucleotides can be up to 10 times greater than the chosen window size).
- Column 4: Number of reads from the treated sample mapped in the given window.
- Column 5: Number of reads from the control sample mapped in the given window (note that these are raw, not normalized values, so a higher number of reads does not necessarily mean enrichment; use q-values to identify regions statistically enriched in treated sample reads, i.e. "fragile" for DSB sequencing data)
- Column 6: The p-value for the given window is computed using the hypergeometric test. Here, the population size is defined as the sum of the total number of reads in a given chromosome for treated and non-treated samples; the sample size as the number total of reads in the given window for treated and non-treated samples; the success in population as the number of reads in the given chromosome for the treated sample; the sample size is the sum of the number of reads in the given window for treated and non-treated samples; the success in the sample is defined as the number of reads in a given window for the treated sample.
- Column 7: p-value after Benjamini-Hochberg correction (Q-value #1). Use for samples from organisms with relatively large genomes (e.g. human, mouse).
- Column 8: p-value after Bonferroni correction (Q-value #2). Use for samples from organisms with relatively small genomes (e.g. yeast).
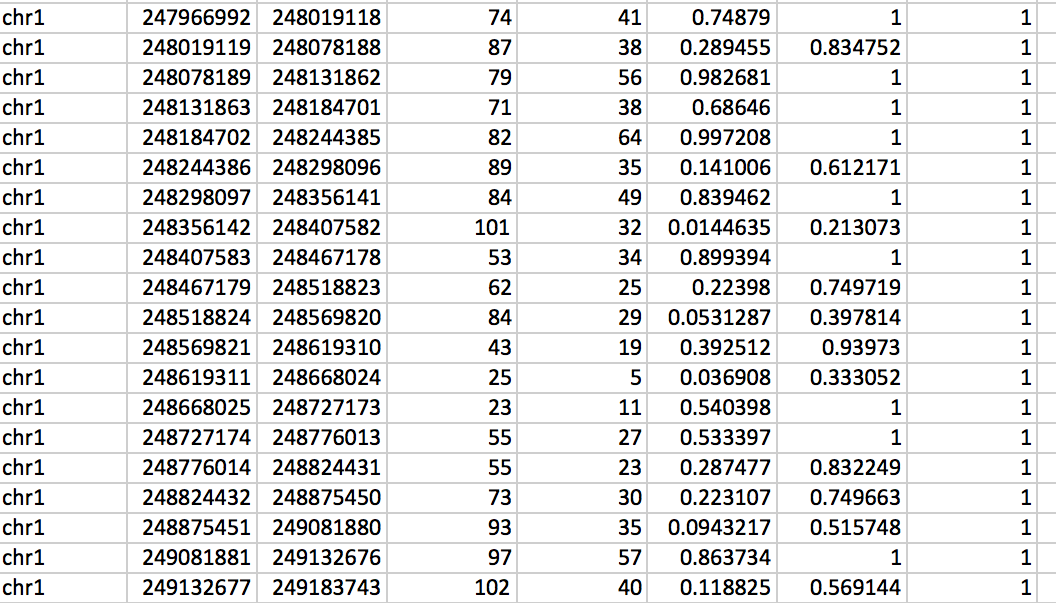
Table 1: Example of hygestat_BLESS output template
- Annotating the results
Hygestat_annotation annotates enriched regions found by hygestat_BLESS using genome annotation data. Program input consists of the following files:
- ygestat_BLESS output (or any data file in compatible format,containing some genomic features binned into equal size (or equal size in mappable nucleotides) genomic windows),
- annotation file in BED or bedGraph format, containing any discrete or continuous genomic annotations, respectively,
- (optional) genome mappability data, imported to bitarray format.
Options
Definitions:Option Extended option Argument / Usage Description Default argument -h ---help show the help message and exit -F --feature-file FFILE / --feature-file=FFILE path to the annotation data file [no default] -B --bless-file BFILE / --bless-file=BFILE path to the bless data file [no default] -b --bless-name BNAME / --bless-name=BNAME BLESS experiment name for output data [default: path to the bless data file] -f --feature-name FNAME / --feature-name=FNAME annotation name for output data [default: path to the annotation data file] -g --bedgraph produces annotation data in bedGraph format [default: BED format] -O --output-file OUTFILE / --output-file=OUTFILE path to the output file [no default] -a --append-output appends output to existing file [default: create a new file] -p --pvalue-thresholds PVTHRS / --pvalue-thresholds=PVTHRS comma separated list of p-value thresholds for BLESS data [default: p-value threshold is 0.05] -c --pvalue-column PVCOL / --pvalue-column=PVCOL p-value column in bless data file [default: 7] -M --mappability-dir MAPDIR / --mappability-dir=MAPDIR path to the mappability data directory [default:no mappability] -n --n-tests NTEST / --n-tests=NTEST number of permutation tests [default: 1000 ] - FFILE: annotation data in BED or bedGraph format to be compared with BLESS data. All but the first three (BED) or four (bedGraph) fields in each line are ignored.
- BFILE: BLESS data output from hygestat_BLESS.
- FNAME and BNAME: input data names used in the output file (see OUTPUT FORMAT below).
- OUTFILE: output is written as a tab-separated text file (see OUTPUT FORMAT below).
- PVTHRS: significance level thresholds for BLESS data.
- PVCOL: column determining p-value type for BLESS data; possible values are:
- p-value from the hypergeometric test,
- Benjamini-Hochberg corrected hypergeometric p-value (default),
- Bonferroni corrected hypergeometric p-value.
- MAPDIR: directory with the mappability data imported to binary format (see MAPPABILITY below);
- NTEST: the number of permutations in the enrichment/depletion estimation; resulting p-value resolution equals 1/NTEST.
- feature - FNAME argument
- treatment - BNAME argument
- pv_threshold - BLESS p-value threshold, a component of PVTHRS argument
- level_all - the fraction of nucleotides overlapping with the selected feature in the genome
- level_fragile - the fraction of nucleotides overlapping with the selected feature within hygestat_BLESS-significant windows (or within window p-value <= pv_threshold if non-default of PVTHRS is selected)
- level_proportion - level_proportion - enrichment of the selected feature in hygestat_BLESS significant ("fragile") intervals (level_fragile/level_all ratio)
- pval_enrichment - p-value for the enrichment (level_proportion)
- pval_depletion - p-value for the depletion (level_proportion) .
- Analyzing enrichments in genes (hygestat_genes)
hygestat_genes works as hygestat_BLESS (described above), but instead of using same size mappable windows for binning the data, it will assign reads to genes. To run hygestat_genes:
- Download the zipped package from our website. After unzipping the package (
$unzip Hygestat_genes) type$cd Hygestat_genesto change directory and then $make to compile the program and obtain the executable file (hygestat_genes). - Perform the command $./hygestat_genes treated_xx_barcode/ untreated_xx_barcode/ mappability_directory/ gene_code_annotation.gft, which executes the program.
- Chromosome number
- Gene name
- Gene start position
- Gene end position
- Gene size
- Mappable length
- Number of reads in treatment (treated sample) for the given gene.
- Number of reads in control (untreated sample) for the given gene.
- P-value for the given gene being enriched in breaks (or other measured characteristic) (hypergeometric test).
- Q-value, i.e. p-value corrected for multiple hypothesis testing (Benjamini-Hochberg correction).
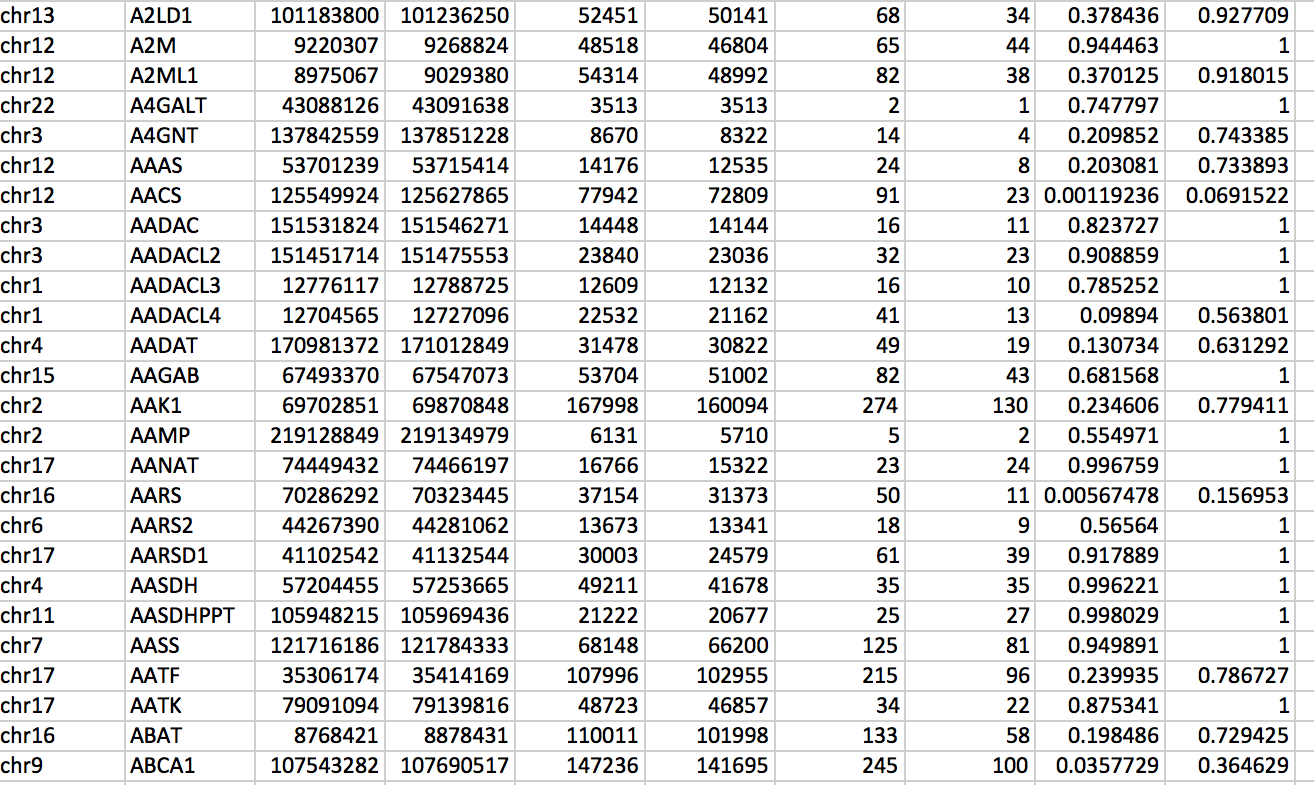
Table 2: Example of an output file produced by hygestat_genes.
- Download the zipped package from our website. After unzipping the package (
- Analyzing enrichments in user-provided intervals (hygestat_bed)
Hygestat_bed uses custom regions, provided by the user, to evaluate enrichment of DNA breaks (or other measured feature), in these regions.
To use, download the zipped package called Hygestat_bed.zip from our website. After unzipping the package ($unzip Hygestat_bed.zip), type $cd Hygestat_bed
to change the directory and $make to compile the program and obtain the executable file (hygestat_bed).
Use the following command to execute the program: $./hygestat_bed treated_xx_barcode untreated_xx_barcode size_window bed_regions
Notes:treated_xx_barcode and untreated_xx_barcode are treated and untreated samples (as in hygestat_BLESS).
A bed_file is a text file with three or more tab separated columns that contain the chromosome numbers and genomic addresses (begin and end coordinates) of some genomic regions of interest.
Fourth and further columns of a bed file are ignored by hygestat_bed.
Additionally, an option specifying minimum window size to be accepted among those provided in the
bed file can be used (default is 1, that all windows in the user-provided bed file are analyzed):
$./hygestat_bed treated_xx_barcode untreated_xx_barcode size_window bed_regions.
The output of hygestat_bed (see also Table 3) contains the following eleven columns:
- Chromosome number (chrXX)
- 2. Window beginning address
- 3. Window end address
- Window size
- 5. Number of treatment reads in the given window
- 6. Number of control reads in the given window
- 7. P-value for the given window to be enriched (obtained from the hypergeometric test)
- 8. Q-value1 (calculated using Benjamini Hochberg multiple hypothesis testing correction)
- 9. Q-value2 (calculated using Bonferroni multiple testing correction. Bonferroni method yields more conservative q-values than the Benjamini-Hochberg method and is thus recommended for deeply sequenced samples, where sensitivity of the detection is not a concern)
- -Log of p-value
- -Log of q-value1
- -Log of q-value2
Table 3: Example of hygestat_bed output table
- Analyzing locations of significant regions with respect to transcription start and end sites (find_gene)
This function uses the hygestat_BLESS (or hygestat_bed) output to find the nearest genes for each region.
The user can determine in which distance from transcription start sites or ends to search for enriched intervals found by other hygestat_programs.
The software also returns the orientation of the found fragment vs. gene. For example, whether an enriched region was found upstream/in the gene promoter (user can define distance cut-off),
within a gene body, or downstream. This functionality can be used for example to analyzing if DSBs are enriched in promoters of differentially expressed genes, as we did in (cite).
It can be obtained from our website (breakome/software) and it works as follow: Download the zip package from our website and after unzipping it,
type $cd Find_gene to change the directory and $make to get the executable file.
To execute the program, use the following command: $./find_gene hygestat_window_output.txt gene_code_annotation.gtf distance_parameter p-value_threshold.
The output file (Table 4) can be found in output.txt and is interpreted as follow:
- Chromosome name (chrXX)
- Gene name
- Gene start position
- Gene end position
- Strand
- Window start position
- Window end position
- Distance from gene
- Gene position
- P-value
- Distance parameter
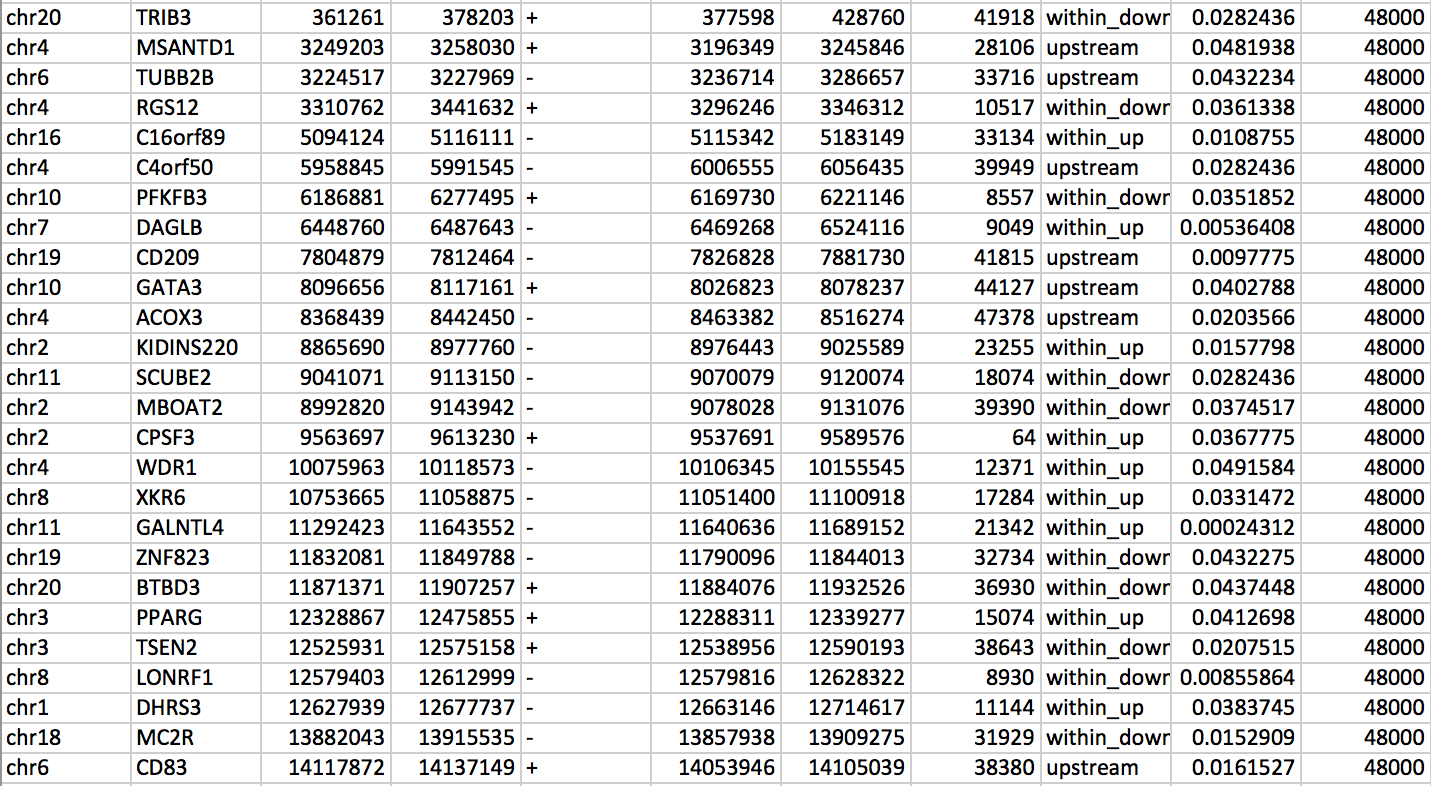
Table 4: Eleven columns table results showing an overview of the find genes program output
- Visualizing the data on the chromosomes using custom Matlab code The pipeline software for visualization is available on our website to be downloaded (breakome). Follow the steps on the readme file to obtain the plots.
- TROUBLESHOOTING
- TIMING
The computational time for the whole analysis entirely depends on the data set and size, however:
- Steps 1 – 3 read the data, extracting reads, and removing barcodes takes less then 10min
- Step 4, aligning fragment reads to the reference genome takes less than 35min
- Step 5, Reading bowtie output and extracting mapped position take less than 3min
- Step 6, Reading of pre-computed mappable regions and finding reads overlapped with those mappable regions. This process takes less than 5min
- Steps 7-10, evaluate p-values for each region and writing the output file, this process takes less than 5min
- Step 11, finding the annotation of the data with the well-known data from genome browser (NCBI) takes about 30 min depending of the precision of the results
- Step 12, Running hygestat genes function to find gene prone fragility takes no longer than 10min
- Step 13, Running hygestat bed regions to evaluate the probability of given regions of being fragile or double strand breaks (DSBs) prone regions. This process takes less than 10 min
- Step 14, Finding the surrounding genes of the region of interest by running find genes function. This process takes no longer than 1h depending of the distance parameter (distance from the region of interest)
Based on the advanced with computational tools, today’s computational timing should not be the limit for the high quality of analysis. We test the computational timing for the whole data set from Crosetto et al. during this protocol. Running the whole process in a DELL machine (characteristics) for hygestat window takes no longer than 40min. The most timing process is to align the reads to the reference genome. As others software use the pre-processing data from hygestat window (mapped data and hygestat window output), the computational time is considerably reduced and vary from 2min to 10min.
- ANTICIPATED RESULTS
In this section, we applied each step described above to the data from Crosetto et al. and recovered the results as described in the publication.
The data can be obtained under accession number SRP018506 or from our website (breakome).
Following the steps given above (procedure), we show in this section the distribution of DNA double strand breaks (DSBs) across each chromosome as described in the original paper (Crosetto et al.).
Figure 3 bellow presents the regions prone DSBs for each chromosome.
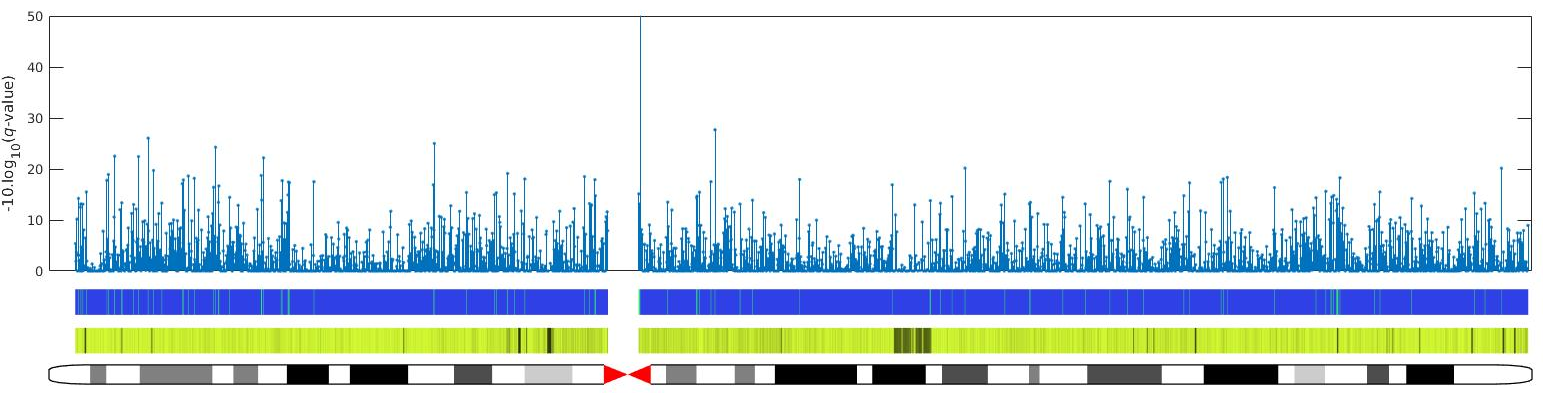
Fig 3: An example of chromosome X (human genome) using data from Nature Method (Crosetto et all.) to show the distribution of DSBs across the chromosome. The top panel represents the -10log(q-val) for each region on the chromosome with 48k window size. In the second panel from the top, the green bars represent the regions prone DSBs. The third panel represents 48kbp mappability region with black bars as non-mappable regions.
How to cite iSeq
Publications using iSeq
- Crosetto N., Mitra A., Silva M. J. et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nature Methods 10, 361–365 (2013)
- Abhishek Mitra, Magdalena Skrzypczak, Krzysztof Ginalski, Maga Rowicka. Strategies for Achieving High Sequencing Accuracy for Low Diversity Samples and Avoiding Sample Bleeding Using Illumina Platform. PLoS ONE 10(4): e0120520. doi:10.1371/journal.pone.0120520 (2015)
- Yang J, Mitra A, Dojer N, Fu S, Rowicka M, Brasier AR. A probabilistic approach to learn chromatin architecture and accurate inference of the NF-κB/RelA regulatory network using ChIP-Seq. Nucl. Acids Res. 41 (15): 7240-7259 (2013).
- Xueling Li, Yingxin Zhao, Bing Tian, Mohammad Jamaluddin, Abhishek Mitra, Jun Yang et al. Modulation of Gene Expression Regulated by the Transcription Factor NF-κB/RelA The Journal of Biological Chemistry 289, 11927-11944 (2014).
- W. Shi, T. Vu, D. Boucher, A. Biernacka, J. Nde, R. K. Pandita et al. Ssb1 and Ssb2 have essential and overlapping functions in self-renewing organ homeostasis. Blood (2017).
- F. Aymard, M. Aguirrebengoa, E. Guillou, B. Maria-Javierre, B. Bugler, C. Arnould et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat Struct Mol Biol. 2017 Apr;24(4):353-361. doi: 10.1038/nsmb.3387